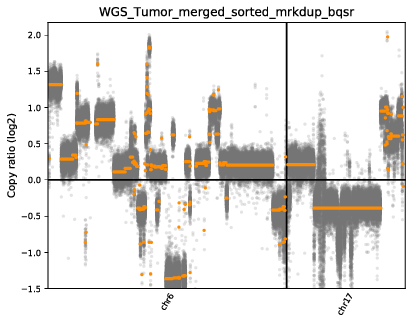
Run CNVkit to identify CNV
July 23, 2024
It is easy to find a pipeline and call for results. However, how do you confirm the results are valid? One of the good approaches is to reproduce tutorials.
Goal
This [1] uses human genome to identify CNV using cnvkit [2]. We will reproduce the tutorial's analysis by downloading their raw data, running the analysis, and comparing the results with their provided outcome to better understand the pipeline.
Download data
mkdir data
cd data
# Get ref.fa
wget -c http://genomedata.org/pmbio-workshop/references/genome/all/ref_genome.tar
tar -xvf ref_genome.targunzip
gunzip ref_genome.fa.gz
# Get copyCat bed
wget -c http://genomedata.org/pmbio-workshop/misc/copyCat_annotation.zip
unzip copyCat_annotation.zip
# Get Exom target bed
wget -c http://genomedata.org/pmbio-workshop/references/exome/SeqCapEZ_Exome_v3.0_Design_Annotation_files.zip
unzip SeqCapEZ_Exome_v3.0_Design_Annotation_files.zip
# Get bam
wget -r -np -nH --cut-dirs=4 -R "index.html*" http://genomedata.org/pmbio-workshop/results/chr6_and_chr17/align/
# Get wgs bed
wget http://genomedata.org/pmbio-workshop/results/chr6_and_chr17/somatic/cnvkit_wgs/access-excludes.hg38.chr6_and_17.target.bed
# Get results of only chr6 and chr17
cd results
wget -r -np -nH --cut-dirs=4 -R "index.html*" http://genomedata.org/pmbio-workshop/results/chr6_and_chr17/somatic/cnvkit_exome/
# Get results of wgs
wget -r -np -nH --cut-dirs=4 -R "index.html*" http://genomedata.org/pmbio-workshop/results/chr6_and_chr17/somatic/cnvkit_wgs/
Run analysis
Prepare path
conda activate py38_cnvkit
export PYTHONPATH=PATH_TO_PYTHON
# Path
cnvkit_path="python -m cnvkit.cnvlib.cnvkit "
WD=WORK_DIR/
input_path=$WD/data
REF=$input_path/ref_genome.fa
copyCat_annotation_bed=$input_path/copyCat_annotation/gaps.bed
output_path=$WD/sandbox
mkdir -p $output_path
Run analysis
###### Prepare data
# Calculate the regions of the genome which are inaccessible to sequencing
$cnvkit_path access $REF -x $copyCat_annotation_bed -o $output_path/access-excludes.hg38.bed
# cnvkit will complain if access-excludes contains chromosomes not in the bam file
# we subset to chr6 and chr17 here to avoid this error later
grep "chr6\|chr17" $output_path/access-excludes.hg38.bed > $output_path/access-excludes.hg38.chr6_and_17.bed
####### Run analysis
# cnvkit will complain if chromosomes are in the bed file but not the bam, we fix this here
grep "chr6\|chr17" $input_path/SeqCapEZ_Exome_v3.0_Design_Annotation_files/SeqCap_EZ_Exome_v3_hg19_primary_targets.bed > $input_path/SeqCap_EZ_Exome_v3_hg38_primary_targets.v2.chr6_and_17.bed
# run the entire cnvkit workflow for the exome data
$cnvkit_path batch $input_path/Exome_Tumor_sorted_mrkdup_bqsr.bam --normal $input_path/Exome_Norm_sorted_mrkdup_bqsr.bam --targets $input_path/SeqCap_EZ_Exome_v3_hg38_primary_targets.v2.chr6_and_17.bed --fasta $REF --access $output_path/access-excludes.hg38.chr6_and_17.bed --output-reference $output_path/my_reference.cnn --output-dir $output_path --method hybrid -p 8 --diagram --scatter --drop-low-coverage
## Run whole genome seq
# make directory to store results
mkdir -p $output_path/cnvkit_wgs
cd $output_path/cnvkit_wgs
# run the cnvkit pipeline
$cnvkit_path batch $input_path/WGS_Tumor_merged_sorted_mrkdup_bqsr.bam --normal $input_path/WGS_Norm_merged_sorted_mrkdup_bqsr.bam --fasta $REF --access $input_path/access-excludes.hg38.chr6_and_17.target.bed --output-reference $output_path/cnvkit_wgs/my_reference.cnn --output-dir $output_path/cnvkit_wgs --method wgs -p 8 --diagram --scatter
Results
There are files and plots results that we can use to compare. And we successfully replicated the tutorial.