
Quality control and trim for fastqs
Before starting aligning the fastqs to the reference genome, we need to do the important step of quality control. Here we use two tools, Fastqc [1] as a quality control tool for high throughput sequence data and fastp [2] as a comprehensive tool for filtering bad reads and trimming adapter.
0. Prepare the fastqs
When samples were sent for sequencing, they may be labeled differently from their original sample name. To keep our analysis clear, let's first rename all fastqs with their sample names.
List files with paths in Linux
Full path
find "$PWD"
Relative path
find .
# Create the dict with format sample name:lable name
find . | grep "fq.gz" | sed 's=./==;s=_1.fq.gz==;s=_2.fq.gz==;' | sed 's=/=:=' | sort | uniq > sample_label_dict.txt
mkdir raw_fq
WD=WORK_DIR_TO_THE_FILES
out=$WD/raw_fq
for i in $(cat sample_label_dict.txt)
do
sample=$(echo $i | cut -d ":" -f 1)
label=$(echo $i | cut -d ":" -f 2)
ln -s $WD/$sample/$label\_1.fq.gz $out/$sample\_1.fq.gz
ln -s $WD/$sample/$label\_2.fq.gz $out/$sample\_2.fq.gz
done
1. Use FastQC to see the original quality of fastqs
#!/bin/bash
mkdir fastQC
mkdir Shfiles
mkdir Log
FQ_DB=raw_fq
for i in $(ls $FQ_DB/* );
do SAMPLE=$(echo $i|sed 's=raw_fq/==;s/.fq.gz//')
sed "s/Hi/$SAMPLE\_QC/" Model.sh > Shfiles/$SAMPLE\_QC.sh
echo fastqc -o fastQC -t 7 $i >> Shfiles/$SAMPLE\_QC.sh
sbatch Shfiles/$SAMPLE\_QC.sh
done
1.5 Summarize results from FastQC
Copyright: Karobben's bolg: https://karobben.github.io/
This part was created by my excellent colleague Karobben [3]. In the result html of FastQC, we can see a graph like this [1:1]:
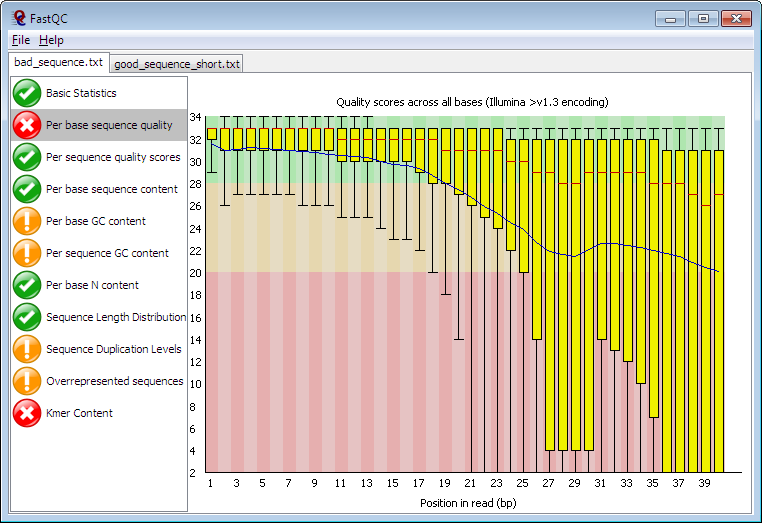 Karobeen implemented tools like web scrapping, R, and Python to save our time of looking at individual quality control html result produced by
Karobeen implemented tools like web scrapping, R, and Python to save our time of looking at individual quality control html result produced by FastQC.
A. Create a csv storing the information
import os
import pandas as pd
from bs4 import BeautifulSoup
def Tab_grep(Sample):
html = open(Sample).read()
soup = BeautifulSoup(html, features='lxml')
Summary = soup.find_all('div',{"class":"summary"})[0]
Reu_l = [Sample]
Cla_l = ["Sample"]
for line in Summary.find_all("li"):
Cla_l += [line.get_text()]
Reu_l += [str(line).split('"')[1]]
Result_TB = pd.DataFrame([Reu_l], columns=Cla_l)
return Result_TB
Result_TB = pd.DataFrame()
for Sample in [i for i in os.listdir() if "fastqc.html" in i]:
Result_TB = pd.concat([Result_TB, Tab_grep(Sample)])
Result_TB.to_csv("QC.csv")
B. Plot information in R
library(ggplot2)
library(reshape2)
TB <- read.csv("QC.csv")[-1]
TB_P <- melt(TB, id.vars = "Sample")
ggplot() + geom_tile(data= TB_P, aes(Sample,variable, fill= value))
C. Save the per sequence GC content plots in one file
import os
import pandas as pd
from bs4 import BeautifulSoup
def Pic_save(Sample, OUT="/home/wliu15/OUT.md"):
html = open(Sample).read()
soup = BeautifulSoup(html, features='lxml')
F = open(OUT,"a")
F.write(Sample+"\n")
F.write(str(soup.find('h2',{"id":"M5"})))
F.write(str(soup.find('img',{"alt" : "Per base sequence content"})))
F.close()
for Sample in [i for i in os.listdir() if "fastqc.html" in i]:
Pic_save(Sample)
D. Summarize the overrepresented sequences
import pandas as pd
TB = pd.DataFrame()
for Sample in [i for i in os.listdir() if "fastqc.html" in i]:
if len(pd.read_html(Sample))!=1:
TMP = pd.read_html(Sample)[1]
TMP['Sample'] = Sample
TB = pd.concat([TB, TMP])
These sequences can be used to BLAST [4] to determine potential contamination cause.
For how the results look like and more bioinformatics skills, check Karobben's bolg
2. Use fastp to cut bad reads and trim adapters
#!/bin/bash
FQ_DB=raw_fq
mkdir fastp_fq
out=fastp_fq
for i in $(cat sample_label_dict.txt)
do SAMPLE=$(echo $i | cut -d ":" -f 1)
sed "s/Hi/$SAMPLE\_fq/" Model.sh >> Shfiles/$SAMPLE\_fp.sh
echo fastp -i $FQ_DB/$SAMPLE\_1.fq.gz -I $FQ_DB/$SAMPLE\_2.fq.gz -o $out/$i\_1.fq.gz -O $out/$SAMPLE\_2.fq.gz > Shfiles/$SAMPLE\_fp.sh
echo mv fastp.html $out/$SAMPLE\_fastp.html > Shfiles/$SAMPLE\_fp.sh
echo mv fastp.json $out/$SAMPLE\_fastp.json > Shfiles/$SAMPLE\_fp.sh
sbatch Shfiles/$SAMPLE\_fp.sh
done
Supplementary [3:1]
Model.sh
#!/bin/sh
#SBATCH --qos=normal # Quality of Service
#SBATCH --job-name=Hi # Job Name
#SBATCH --time=1-0:00:00 # WallTime
#SBATCH --nodes=1 # Number of Nodes
#SBATCH --ntasks-per-node=1 # Number of tasks (MPI processes)
#SBATCH --cpus-per-task=1 # Number of threads per task (OMP threads)
#SBATCH --output=Log/Hi.out ### File in which to store job output